富山大学は弘前大学と共同で,幼小児期から光線過敏を起こす遺伝性疾患である骨髄性プロトポルフィリン症(Erythropoietic protoporphyria:EPP)の日本人患者の特徴を解析し,日本人に特有な不完全骨髄性プロトポルフィリン症(incomplete EPP)を見出した(ニュースリリース)。
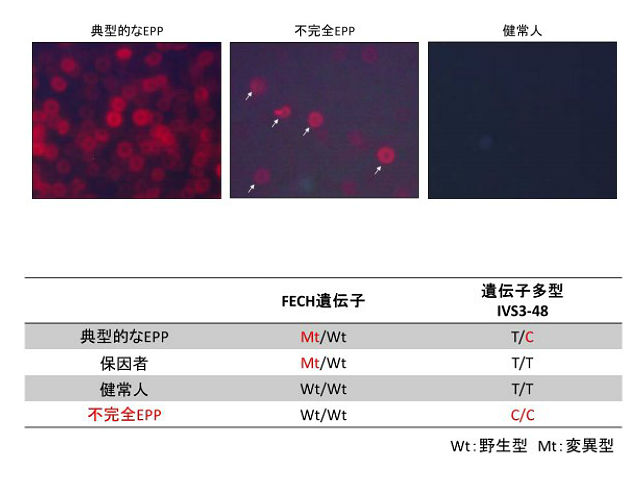
骨髄性プロトポルフィリン症(EPP)は小児期以降に発症する光線過敏を呈する疾患であり,フェロケラターゼ(FECH)をコードするFECH遺伝子が原因遺伝子。ヘムを合成する代謝経路の最終段階において,FECHの活性が低下することによりプロトポルフィリンⅨ(PPIX)が骨髄造血系や皮膚などに蓄積するために生じる。
EPPでは幼少時期から日光曝露後に発赤,疼痛などの光線過敏症状を生じ,時に肝機能障害を起こす。血液検査では蛍光赤血球が出現し,血中プロトポルフィリン(PP)値の高値を呈する。EPPはFECH遺伝子変異単独では発症せず,イントロン3に遺伝子多型IVS3-48T/C(野生型はIVS3-48T/T)を同時に持つことで酵素活性が低下し,EPPが発症することが明らかにされている。
研究グループは,軽度のEPPの所見(軽度の光線過敏症状,少数の蛍光赤血球,血中PP値の軽度上昇)がある3症例を検討したところ,FECH遺伝子の異常は検出されず,遺伝子多型IVS3-48C/Cを同定した。FECH遺伝子における遺伝子多型IVS3-48C/Cにより,FECH活性の減少が引き起こされ,不完全なEPP(incomplete EPP)を発症することを推察した。
さらに,研究グループは,incomplete EPP患者の臨床症状と血中PP値の推移を追った。3症例とも臨床症状は徐々に軽快し,血中PP値の軽度高値は持続したものの,うち1例は正常上限近くまで低下がみられた。このことからincomplete EPPは,成長とともに改善がみられる可能性があると考えられるという。
遺伝子多型IVS3-48C/Cの頻度は人種差があり,日本人では欧米諸国の約10倍多いと考えられている。研究グループは,今回の研究は,incomplete EPPにおいて日本人では遺伝子多型IVS3-48C/Cの頻度が高いことを見出し,これまで診断がつかずに見逃されていた幼児,小児における軽症の光線過敏症が潜在的に存在していることを示唆する世界初の報告としている。